Research Themes
Artificial intelligence (AI) is transforming our approach to molecular engineering. Driven by the goal of accelerating drug development and materials design, our aim is to develop AI-driven methods that enhance how we discover and optimize molecules. We organize our work into two layers. The first is a set of method development pillars, where we build the generative and predictive tools that learn from both structured and unstructured data, spanning molecular structures, chemical reactions, physical simulations, and biomedical data such as single-cell transcriptomics and microscopy images. The second is a set of application domains, where we put those methods to work on concrete therapeutic and materials problems. Increasingly, our methods also draw on chemical language models, foundation models, and agentic workflows to make sense of the unstructured chemical literature and to scale data curation and design.
Method development
Our methods development is organized around six pillars. Together they form a pipeline: from how molecules are represented and the data we learn from, through simulation and accurate property prediction, to the design of new molecules and the prediction of how to make them.
Diverse molecular representations
How a molecule is encoded shapes everything a model can learn from it. We study a wide range of molecular representations, from string and descriptor-based encodings to molecular graphs and higher-order topological representations that capture many-body structure through hypergraph and topological message passing. A recurring question is which representations generalize best across chemical space, whether the target is a small-molecule drug or a copolymer.
Selected publications
- Martínez Crespo, Pablo et al. (2025) “TopoMole: Topological Message Passing Meets Hyperedge Messages.” AI4Mat @ NeurIPS 2025. link
- Kazemi-Khasragh, Elaheh et al. (2026) “Descriptor and Graph-based Molecular Representations in Prediction of Copolymer Properties Using Machine Learning.” Comput. Mater. Sci. link
- David, Laurianne et al. (2020) “Molecular representations in AI-driven drug discovery: a review and practical guide.” J. Cheminf. link
Data mining
Good models need good data, and much of chemistry’s most valuable information sits locked in unstructured text, figures, and tables. We develop methods to extract, structure, and curate chemical data at scale. This includes agentic literature-extraction workflows that augment sparse databases, for example in targeted protein degradation, and tools that identify and split molecular substructures from raw records. We also advocate for community standards that make reaction and assay data reusable in the first place.
Selected publications
- Rao, Yaochen et al. (2026) “Beyond Manual Curation: Augmenting Targeted Protein Degradation Databases via Agentic Literature Extraction Workflows.” arXiv. link
- Mercado, Rocío et al. (2023) “Data sharing in chemistry: lessons learned and a case for mandating structured reaction data.” J. Chem. Inf. Model. link
Simulation for molecular understanding and data generation
Molecular simulations are both a way to understand the interactions and dynamics that govern a biological or chemical process and a way to generate training data where experimental data is scarce. We use classical molecular dynamics (MD) and ab initio methods such as density functional theory (DFT) to study systems of interest and to produce labelled data for predictive models, including unified datasets that combine experiment and simulation paired with fast surrogate models. One current interest is using MD to predict ternary complex formation when designing multi-target therapeutics.
Selected publications
- Beckmann, Richard et al. (2026) “A unified experimental-simulation dataset and surrogate models for surfactant property prediction.” ChemRxiv. link
- Witherspoon, Velencia J. & Mercado, Rocío et al. (2019) “Combined nuclear magnetic resonance and molecular dynamics study of methane adsorption in M2(dobdc) metal–organic frameworks.” J. Phys. Chem. C. 123(19). 12286-12295. link
- Mercado, Rocío et al. (2016) “Force field development from periodic density functional theory calculations for gas separation applications using metal–organic frameworks.” J. Phys. Chem. C. 120(23). 12590-12604. link
Single- and multi-task models for accurate molecular property prediction
Accurate property prediction underpins both ranking candidate molecules and rewarding generative models. We build single- and multi-task models, often sharing information across related endpoints to improve data efficiency, for properties spanning biological activity (e.g., IC50, DC50), ADMET (e.g., permeability, efflux, bioavailability, toxicity), drug metabolism, physico-chemical properties (e.g., pKa, solubility), and ternary complex formation. We pay close attention to evaluation, since headline metrics often mask how a model behaves across chemical space.
Selected publications
- Ohlsson, Philip Ivers et al. (2025) “Prediction of Permeability and Efflux Using Multitask Learning.” ACS Omega. link
- Larsson, Sofia & Carlsson, Miranda et al. (2025) “LAGOM: A Transformer-Based Chemical Language Model for Drug Metabolite Prediction.” AILSCI. link
- Martínez Crespo, Pablo et al. (2026) “QT-Net: Rethinking Evaluation of AI Models in Atomic Chemical Space.” arXiv. link
- Ribes, Stefano et al. (2024) “Modeling PROTAC Degradation Activity with Machine Learning.” AILSCI. link
Molecular design and optimization
At the heart of de novo design is the ability to propose new molecules with a desired property profile and to optimize them efficiently, so as to minimize experimental screening. We develop molecular deep generative models (DGMs) that build molecules in silico, often atom-by-atom or fragment-by-fragment, across a range of therapeutic modalities (Figure 1).
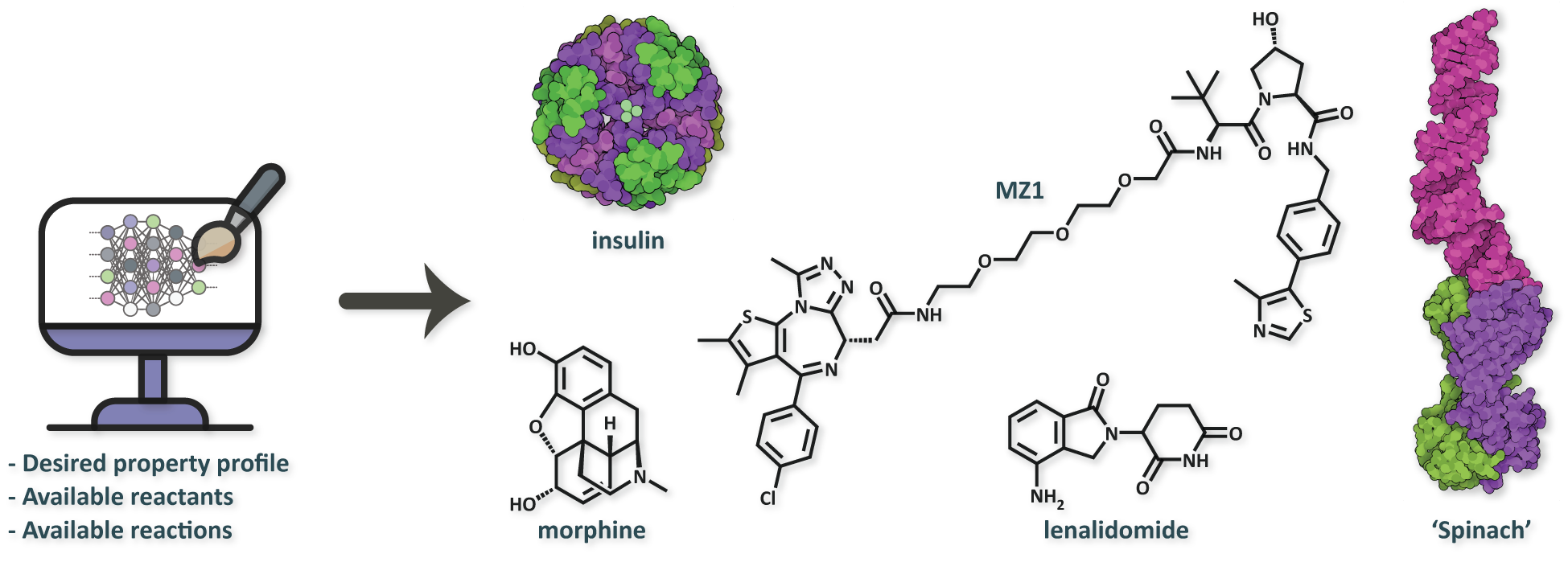
Figure 1. Deep generative models (DGMs) can be developed for a variety of therapeutic modalities. From left to right: a small molecule (agonist), a protein therapeutic, a heterobifunctional degrader, a molecular glue, and an RNA therapeutic (aptamer).
To steer generation toward promising regions of chemical space, we apply reinforcement learning and genetic algorithms, scored by the property prediction models described above. We are also interested in conditional generation, steering models with auxiliary signals such as omics or phenotypic data, and in carefully measuring how well generative models actually cover useful chemical space.
Selected publications
- Romeo Atance, Sara et al. (2022) “De novo drug design using reinforcement learning with graph-based deep generative models.” J. Chem. Inf. Model. link
- Mercado, Rocío et al. (2021) “Exploring graph traversal algorithms in graph-based molecular generation.” J. Chem. Inf. Model. link
- Zhang, Jie et al. (2021) “Comparative study of deep generative models on chemical space coverage.” J. Chem. Inf. Model. link
- Mercado, Rocío et al. (2020) “Graph networks for molecular design.” Mach. Learn.: Sci. Technol. link
Predicting how to synthesize a molecule
A molecule is only useful if it can be made. We develop methods for synthesizability assessment and multi-step retrosynthetic planning, framing route prediction with approaches such as decision transformers and bottom-up synthesis trees. We also study how reliable these models are, including how well large language models agree with human experts on the quality of proposed synthesis plans.
Selected publications
- Granqvist, Emma et al. (2026) “RetroSynFormer: Planning multi-step chemical synthesis routes via a Decision Transformer.” Digital Discovery. link
- Voinarovska, Varvara et al. (2026) “Do humans and large language models agree on the quality of synthesis plans?” ChemRxiv. link
- Gao, Wenhao et al. (2022) “Amortized tree generation for bottom-up synthesis planning and synthesizable molecular design.” ICLR 2022. link
- Westerlund, Annie M. et al. (2024) “Do Chemformers dream of organic matter? Evaluating a transformer model for multi-step retrosynthesis.” J. Chem. Inf. Model. link
Applications
We apply and stress-test these methods in domains where AI-driven design can have outsized impact. Three are central to the group today, alongside emerging directions in materials and life-science discovery.
Targeted protein degradation: PROTACs and molecular glues
Small molecule drugs approved under a “New Drug Application (NDA)” by the FDA comprise only about 80% of the roughly 1,200 new molecular entities approved between 1985 and 2021, with the rest being new biological products. Small molecules (Figure 2) are generally designed to impede the function of biologically-relevant target proteins. A small molecule inhibitor, for instance, binds a protein of interest strongly enough that the protein can no longer carry out its function.
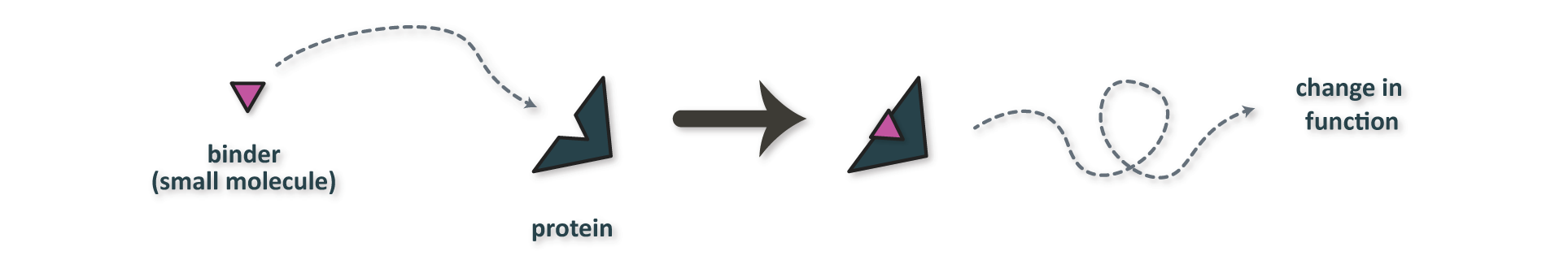
Figure 2. Typical mechanism of action for a small molecule protein binder.
However, an estimated 75% of the human proteome lacks deep binding sites and is considered “undruggable” by traditional small molecule inhibitors. Many of these targets nonetheless drive cancer, autoimmune diseases, and cardio-metabolic diseases, which motivates therapeutic modalities beyond inhibition. We are interested in the controlled design of multi-target modalities such as PROteolysis TArgeting Chimeras (PROTACs; Figure 3) and molecular glues, which can act catalytically at low doses and exploit weak, transient interactions.
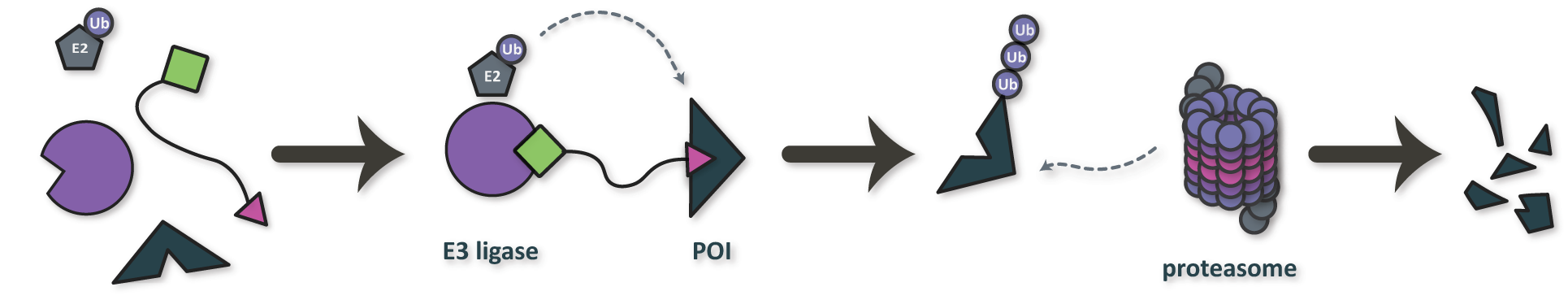
Figure 3. Mechanism of action for a PROteolysis TArgeting Chimera (PROTAC), a class of multi-target therapeutic modality for targeted protein degradation. POI: protein of interest.
A PROTAC uses a three-component structure (Figure 4): a warhead that binds the protein of interest (POI), an E3 ligand that recruits an E3 ligase, and a linker joining them. When the linker holds the two proteins together in a short-lived ternary complex, the POI is ubiquitinated and then degraded by the proteasome. Because the components are highly interdependent, rational design remains challenging, which is why this is one of our most active areas. Our work here draws on every method pillar above: mining and benchmarking degradation data, identifying PROTAC substructures, predicting degradation activity, modeling ternary complex formation with modern structure-prediction tools (such as AlphaFold3 and Boltz-1), and generatively designing degraders.
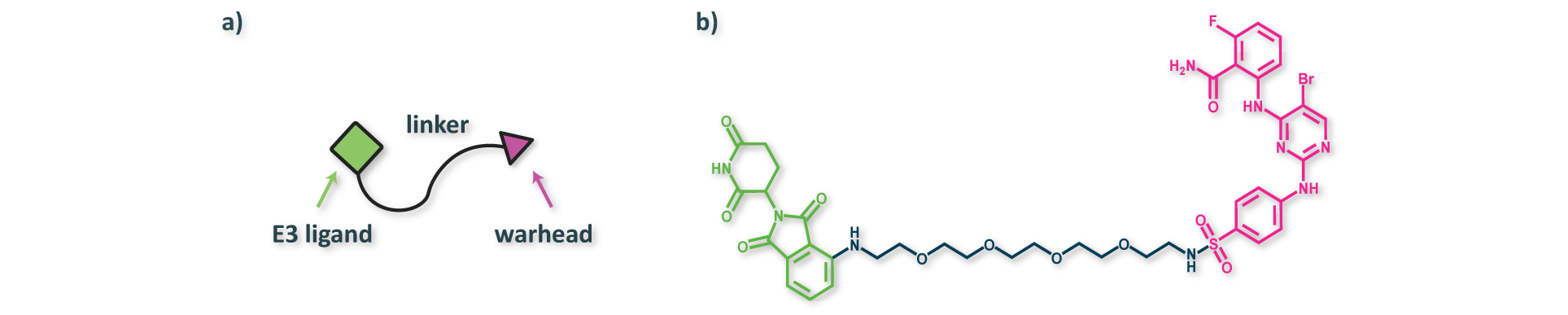
Figure 4. (a) Illustration of the three-component PROTAC structure, and (b) an example PROTAC (PubChem CID: 155168919) from PROTAC-DB. The functionalities of each component are highly interdependent, such that rational design of PROTACs remains challenging.
Selected publications
- Ribes, Stefano et al. (2026) “TACK: A statistical evaluation of degradation activity on a novel TArgeting Chimeras Knowledge dataset.” KDD 2026. link
- Dunlop, Nils & Erazo, Francisco et al. (2025) “Predicting PROTAC-Mediated Ternary Complexes with AlphaFold3 and Boltz-1.” Digital Discovery. link
- Ribes, Stefano et al. (2026) “PROTAC-Splitter: a machine learning framework for automated identification of PROTAC substructures.” J. Cheminf. link
- Gharbi, Yossra et al. (2024) “A Comprehensive Review of Emerging Approaches in Machine Learning for De Novo PROTAC Design.” Digital Discovery. link
- Nori, Divya et al. (2022) “De novo PROTAC design using graph-based deep generative models.” AI4Science @ NeurIPS 2022. link
Solid polymer electrolytes
Safer, higher-performance batteries depend on better electrolytes. In collaboration with researchers at Uppsala University, we apply machine learning to electrolyte modeling and design, including generative design of solvents and the prediction of copolymer properties relevant to solid polymer electrolytes. The goal is to navigate a vast chemical space toward materials that balance ionic conductivity, electrochemical stability, and processability.
Selected publications
- Zhang, Zhan-Yun et al. (2026) “Unlocking the Chemical Space for Rechargeable Batteries with a Generative Solvent Design System.” ChemRxiv. link
- Kazemi-Khasragh, Elaheh et al. (2026) “Descriptor and Graph-based Molecular Representations in Prediction of Copolymer Properties Using Machine Learning.” Comput. Mater. Sci. link
PFAS replacements for semiconductor manufacturing
Many materials used in industrial processes such as semiconductor manufacturing rely on per- and polyfluoroalkyl substances (PFAS), which are persistent and toxic. In collaboration with Intel and EMD Electronics (Merck), we use multi-modal deep learning and generative modeling to discover PFAS alternatives, focusing on the surfactants and photoresists central to chip fabrication. Property prediction from combined experimental and simulated data helps prioritize safer candidates before they are ever synthesized.
Selected publications
- Beckmann, Richard et al. (2026) “A unified experimental-simulation dataset and surrogate models for surfactant property prediction.” ChemRxiv. link
- Mannan, Sajid et al. (2026) “Sustainable Materials Discovery in the Era of Artificial Intelligence.” arXiv. link
Crystalline and inorganic materials discovery
Beyond molecular systems, we work on generative models for crystalline and inorganic materials, and on the benchmarks needed to tell whether such models actually produce useful, synthesizable structures. This includes crystal structure prediction directly from powder diffraction data using autoregressive language models, and contributions to community resources for materials discovery such as LeMaterial.
Selected publications
- Betala, Siddharth et al. (2025) “LeMat-GenBench: A Unified Evaluation Framework for Crystal Generative Models.” arXiv. link
- Lizak Johansen, Frederik et al. (2025) “deCIFer: Crystal Structure Prediction from Powder Diffraction Data using Autoregressive Language Models.” arXiv. link
Phenotype- and omics-guided drug discovery
A growing line of applied work uses high-content biological data, such as microscopy images and single-cell transcriptomics, to guide discovery without relying on a known protein target. We study learned representations of cell images and transcriptomic profiles that support robust phenotypic readouts and, ultimately, phenotype-conditioned molecular design.
Selected publications
- Cropsal, Télio & Mercado, Rocío. (2025) “Compressing Biology: Evaluating the Stable Diffusion VAE for Phenotypic Drug Discovery.” Imageomics @ NeurIPS 2025. link
- Andrekson, Leo et al. (2025) “Contrastive Learning for Robust Cell Annotation and Representation from Single-Cell Transcriptomics.” bioRxiv. link