Research Themes
Artificial intelligence (AI) is transforming our approach to biomolecular engineering. Driven by the goal of accelerating drug development and materials design, our aim is to develop AI-driven molecular engineering methods which will enhance our approach to molecular discovery and optimization. This entails the development of generative and predictive tools that can learn from both structured and unstructured chemical data, such as molecular structures, chemical reactions, and biomedical data.
Deep generative models for molecular engineering
While AI can be applied to a range of molecular engineering tasks, one ideal area is de novo molecular design. De novo design – the concept of designing molecules with desired properties from scratch so as to minimize experimental screening – is poised to allow scientists to more efficiently traverse chemical space in search of optimal molecules, and delegate error-prone decisions to computers via the use of predictive and generative computational models. In drug development, de novo design methods can aid medicinal chemists in the design and selection of drug candidates, with the added advantage that they can learn from datasets of billions of molecules in minutes and be constantly updated with new data. Molecular deep generative models (DGMs) are a particular class of de novo design methods which use deep neural networks to build new molecules in silico, and work by proposing atom-by-atom (or fragment-by-fragment) modifications to an initial graph structure to generate compounds predicted to achieve a certain property profile. Such models can be applied to a range of therapeutic modalities, from small molecules to protein therapeutics (Figure 1).
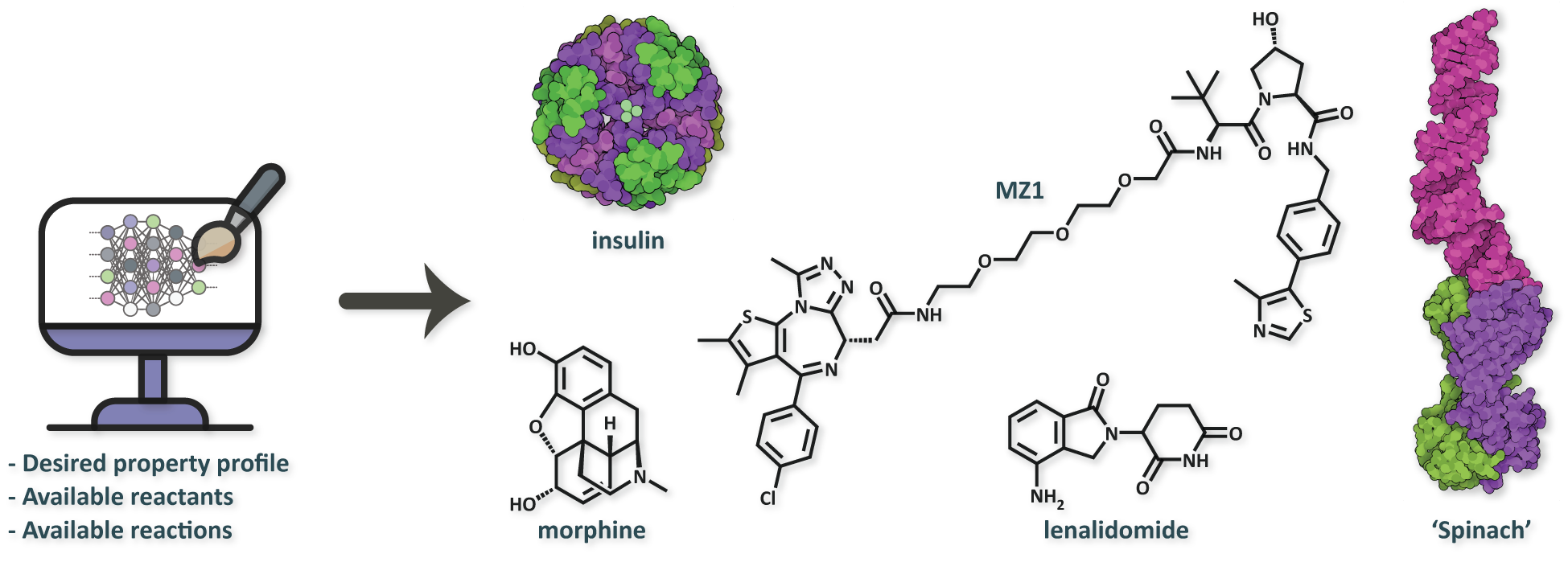
Figure 1. A key research theme in our lab is the development of deep generative models (DGMs) for a variety of therapeutic modalities. From left to right: a small molecule (agonist), a protein therapeutic, a heterobifunctional degrader, a molecular glue, and an RNA therapeutic (aptamer).
Crucial to deep generative models for de novo design is the use of optimization strategies which can guide the model towards promising areas of chemical space. Our group is highly interested in the application of reinforcement learning strategies and genetic algorithms to molecular optimization. A critical component of any optimization strategy is the use of accurate molecular property prediction models which can be used to score or rank molecules. In the cheminformatics literature, these types of models are commonly referred to as quantitative structure-property relationships (QSPR). In the case of policy-gradient reinforcement learning, a property prediction model can be used to reward/punish the reinforcement learning agent, which determines the loss and is used to update the agent parameters such that sampling actions which lead to desirable molecules becomes more likely. The development of improved molecular generative models is inextricably tied to the development of better property prediction models. Properties of interest include biological activity (e.g., IC50, DC50), ADMET (e.g., bioavailability, toxicity), physico-chemical properties (e.g., pKa, solubility), and ternary structure formation.
We are currently interested in applications of deep generative models to the following exciting molecular engineering tasks:
- synthesizability, as ultimately any molecules designed in silico must be synthesized and tested in a lab,
- omics-guided design to leverage existing -omics datasets for the conditional generation of new bioactive molecules,
- and phenotypic screening for the conditional generation of molecules that produce a desired phenotype without any knowledge of the target structure.
Selected publications
- Romeo Atance, Sara et al. (2022) “De novo drug design using reinforcement learning with graph-based deep generative models.” J. Chem. Inf. Model. link
- Gao, Wenhao et al. (2022) “Amortized tree generation for bottom-up synthesis planning and synthesizable molecular design.” ICLR 2022. link
- Mercado, Rocío et al. (2020). “Graph networks for molecular design.” Mach. Learn.: Sci. Technol. link
- Mercado, Rocío et al. (2018). “In silico design of 2D and 3D covalent organic frameworks for methane storage applications.” Chem. Mater. 30(15). 5069-5086. link
Multi-target therapeutic modalities
Small molecule drugs approved under a “New Drug Application (NDA)” by the FDA comprise only ~80% of the 1,200 new molecular entities approved between 1985-2021, with the other 20% being new biological products. Small molecules (Figure 2) are generally designed to impede the function of biologically-relevant target proteins; for instance, small molecule inhibitors interfere with their targets by binding strongly enough to a protein of interest such that it’s behavior is affected and it can no longer carry out it’s function.
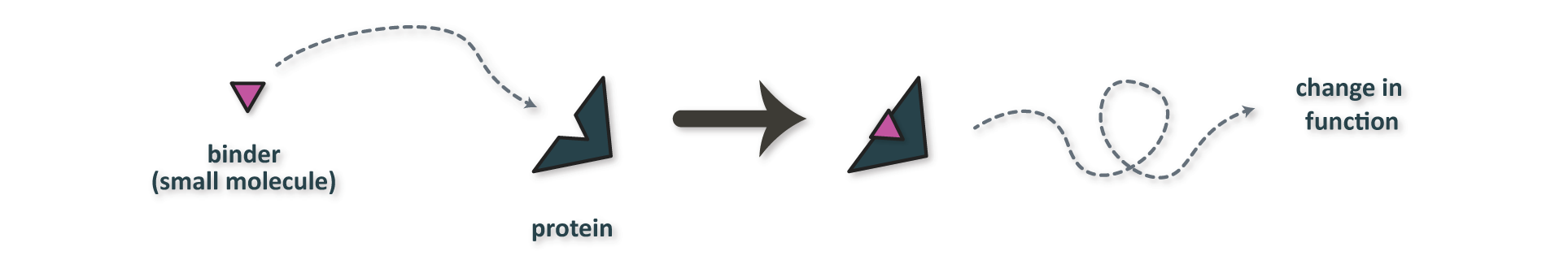
Figure 2. Typical mechanism of action for a small molecule protein binder.
However, it is estimated that ∼75% of the human proteome lacks deep binding sites and is considered “undruggable” by traditional small molecule inhibitors. Nonetheless, these so-called undruggable targets are implicated in a wide range of diseases, including cancer, autoimmune diseases, and cardio-metabolomic diseases, motivating the development of therapeutic modalities beyond small molecule inhibitors. To this end, our group is interested in the development of tools for the controlled design of multi-target therapeutic modalities such as PROteolysis TArgeting Chimeras (PROTACs; Figure 3) and molecular glues.
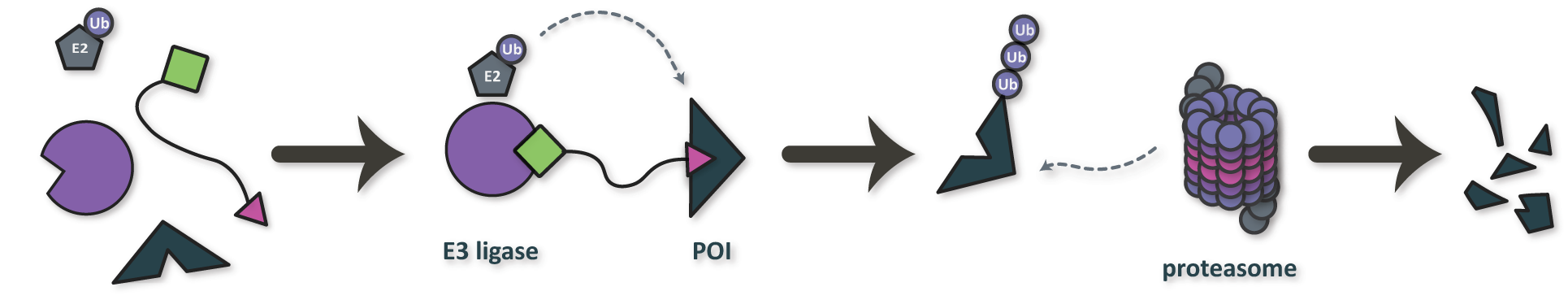
Figure 3. Mechanism of action for a PROteolysis TArgeting Chimera (PROTAC), a class of multi-target therapeutic modality for targeted protein degradation. POI: protein of interest.
The function of PROTACs is generally enabled by a three-component structure consisting of two binding domains and an organic linker (Figure 4). The two binding domains include a warhead designed to bind a protein of interest (POI) and an E3 ligand designed to bind an E3 ligase. Deep generative models can be used to optimize each component independently or to enginere all components of the structure simultaneously. In the ideal scenario, the linker anchors the two proteins together in a short-lived ternary complex, leading to ubiquitination of the POI. The ubiquinated POI is eventually degraded by the proteasome, thus preventing it from carrying out its function in a cell.
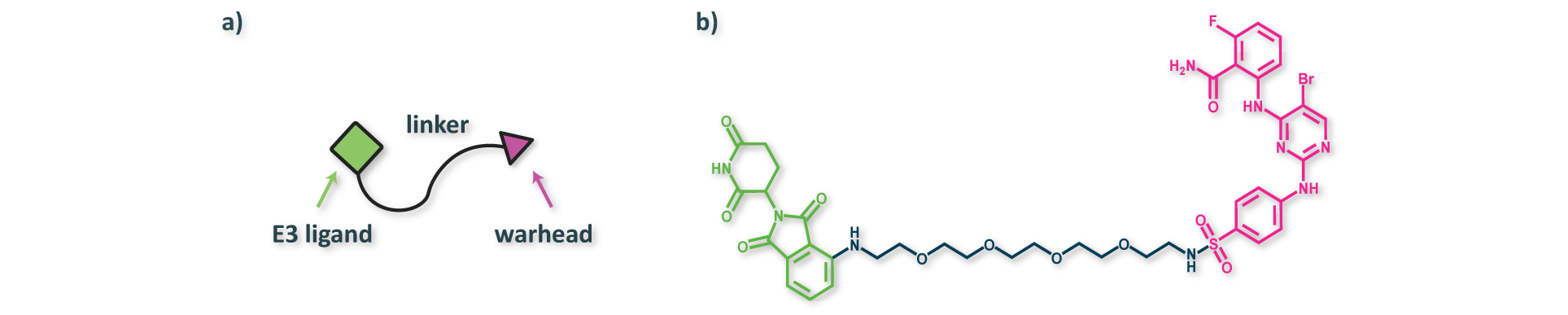
Figure 4. (a) Illustration of the three-component PROTAC structure, and (b) an example PROTAC (PubChem CID: 155168919) from PROTAC-DB. The functionalities of each component are highly interdependent, such that rational design of PROTACs remains challenging.
Not only do such therapeutic modalities open up the space of druggable targets using small molecules, but they can also leverage weak binding interactions with proteins and have a catalytic mechanism of action, requiring lower potential doses in patients. Polypharmacology-based therapeutic approaches are also relevant in the development of treatments of various central nervous system (CNS) disorders, such as Parkinson’s disease, schizophrenia, and substance abuse/addiction. Our group is interested in using computational tools to better understand the biology of these diseases and their potential treatment via multi-target therapeutics. By understanding how a small bound molecule affects a given protein’s interactome, the rational design of targeted protein degraders becomes more tractable.
Selected publications
- Gharbi, Yossra et al. (2024) “A Comprehensive Review of Emerging Approaches in Machine Learning for De Novo PROTAC Design.” arXiv. link
- Nori, Divya et al. (2022) “De novo PROTAC design using graph-based deep generative models.” NeurIPS 2022 AI4Science Workshop. link
Atomistic simulations for molecular understanding and data generation
Molecular simulations are not just a useful tool for gaining a better understanding of the molecular interactions and dynamics which govern a specific biological process, but also a useful tool for data generation. In cases where there is not sufficient data available for developing a predictive model, suitable training data can be generated via computational chemistry methods, such as ab initio calculations and classical molecular simulations. We are currently interested in the use of molecular dynamics simulations for predicting successful ternary complex formation when designing new multi-target therapeutics; such data can in turn be used to predict the extent of protein degradation in a new targeted protein degradation system.
Selected publications
- Witherspoon, Velencia J. & Mercado, Rocío et al. (2019). “Combined nuclear magnetic resonance and molecular dynamics study of methane adsorption in M2(dobdc) metal–organic frameworks.” J. Phys. Chem. C. 123(19). 12286-12295. link
- Mercado, Rocío et al. (2016). “Force field development from periodic density functional theory calculations for gas separation applications using metal–organic frameworks.” J. Phys. Chem. C. 120(23). 12590-12604. link
Sustainable materials discovery
Many materials currently used in industrial processes, such as semiconductor manufacturing, consist of environmentally harmful and toxic chemicals such as per- and polyfluoroalkyl substances (PFAS). As part of a collaboration with Intel and EMD Electronics (Merck), we are interested in using multi-modal deep learning and generative modeling to discover alternatives to these toxic substances. In another collaboration with researchers at Uppsala University, we are exploring the use of machine learning for electrolyte modeling and design.
Selected publications
- Mercado, Rocío et al. (2018). “In silico design of 2D and 3D covalent organic frameworks for methane storage applications.” Chem. Mater. 30(15). 5069-5086. link
- Simon, Cory M. et al. (2015). “The materials genome in action: identifying the performance limits for methane storage.” Energy Environ. Sci. 8. 1190-1199. link